.+Photo+Author.jpg)


ALTERACIONES CONGENITAS DE LA COAGULACION. Expone :DR. VZ Mechan Mendez.

{kind=link}
HEMOFILIAS
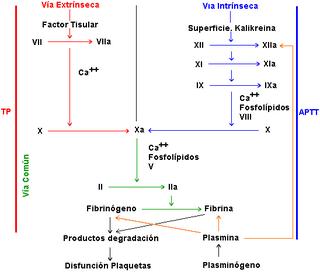
-Hemofilia A (Déficit de F:VIII, herencia ligada al Cromosoma X. Incidencia:1/10 000).
-Hemofilia B (Déficit de F: IX, herencia ligada al C. X. Incidencia 1/60 000).
-Hemofilia C (Déficit de F: XI. Se transmite con carácter autosómico recesivo incompleto afectando a varones y mujeres. Incidencia 1/1 000 000).
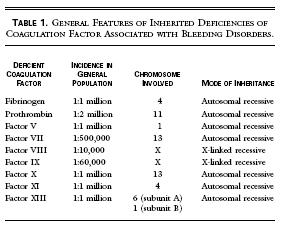 Las Hemofilias A, B y C, son producidas por deficiencias hereditarias (disminución de la producción o, pérdida de la actividad funcional del : F:VIII:C, IX:C u, XI:C, respectivamente. Un 30% de las mutaciones surgen de novo. Tipicamente, el FIXa, activa al FX y el FVIII, forma un complejo con el FIXa, en la superficie de las plaquetas, acelerando la velocidad de activacion del FX, mediante el FIXa. En pacientes con Hemofilia, la formación del coágulo se retrasa debido a una notable disminución de la generación de trombina. El coágulo que se forma es inefectivo, produciendose la hemorragia. Como las deficiencias de FVIII y FIX incapacitan la actividad del FX, ostentan cuadros clinicos similares.
Las Hemofilias A, B y C, son producidas por deficiencias hereditarias (disminución de la producción o, pérdida de la actividad funcional del : F:VIII:C, IX:C u, XI:C, respectivamente. Un 30% de las mutaciones surgen de novo. Tipicamente, el FIXa, activa al FX y el FVIII, forma un complejo con el FIXa, en la superficie de las plaquetas, acelerando la velocidad de activacion del FX, mediante el FIXa. En pacientes con Hemofilia, la formación del coágulo se retrasa debido a una notable disminución de la generación de trombina. El coágulo que se forma es inefectivo, produciendose la hemorragia. Como las deficiencias de FVIII y FIX incapacitan la actividad del FX, ostentan cuadros clinicos similares.
CLINICA DE LA HEMOFILIA A:
Es importante recordar que la síntesis del factor VIII, se realiza fundamentalmente en las celulas endoteliales de los sinusoides hepáticos y en menor cuantia en otros órganos. La hemostasia es normal, con niveles del factor VIII, superiores al 30%. Los niveles de FVIII se mantienen constantes a lo largo de la vida del paciente y son similares entre los miembros de una misma familia, aunque varian entre familias distintas.
Hemartrosis. (Hemorragias intraarticulares). Constituyen el 75% de los episodios hemorrágicos en pacientes con HA, severa. Lugares de ocurrencia frecuente, son : rodillas, codos, tobillos, hombros, muñecas, caderas. Las hemorragias se anuncian con dolor, continuadas con inflamación, incremento de la temperatura articular y limitación de los movimientos. Si hay fiebre persistente sospechar infección. El sangrado recidivante en una articulación produce hipertrofia sinovial e inflamación con limitación del movimiento y tendencia a más sangrado en esa articulación, causando destrucción del cartílago articular, hiperplasia sinovial y deformidad articular con atrofia muscular y contractura de tejidos blandos.
Hematomas : Generalmente sangrado en musculos o tejidos subcutáneos, hematomas intramusculares, en muslos, glúteos, pantorrillas y antebrazos. Se pueden reabsorber, pero tambien aumentar y causar comprensión de organos, nervios, vasos sanguineos, vias aéreas grandes, hematomas encapsulados (pseudotumores organizados, óseos de Volkmann), que comprimen estructuras vecinas. La principal causa de muerte sigue siendo una hemorragia en el SNC. Todas las hemofilias severas cursan con hematurias asociadas a cólicos renales. Las extracciones dentarias, pueden condicionar hemorragias prolongadas.
-Hemofilia A (Déficit de F:VIII, herencia ligada al Cromosoma X. Incidencia:1/10 000).
-Hemofilia B (Déficit de F: IX, herencia ligada al C. X. Incidencia 1/60 000).
-Hemofilia C (Déficit de F: XI. Se transmite con carácter autosómico recesivo incompleto afectando a varones y mujeres. Incidencia 1/1 000 000).
 Las Hemofilias A, B y C, son producidas por deficiencias hereditarias (disminución de la producción o, pérdida de la actividad funcional del : F:VIII:C, IX:C u, XI:C, respectivamente. Un 30% de las mutaciones surgen de novo. Tipicamente, el FIXa, activa al FX y el FVIII, forma un complejo con el FIXa, en la superficie de las plaquetas, acelerando la velocidad de activacion del FX, mediante el FIXa. En pacientes con Hemofilia, la formación del coágulo se retrasa debido a una notable disminución de la generación de trombina. El coágulo que se forma es inefectivo, produciendose la hemorragia. Como las deficiencias de FVIII y FIX incapacitan la actividad del FX, ostentan cuadros clinicos similares.
Las Hemofilias A, B y C, son producidas por deficiencias hereditarias (disminución de la producción o, pérdida de la actividad funcional del : F:VIII:C, IX:C u, XI:C, respectivamente. Un 30% de las mutaciones surgen de novo. Tipicamente, el FIXa, activa al FX y el FVIII, forma un complejo con el FIXa, en la superficie de las plaquetas, acelerando la velocidad de activacion del FX, mediante el FIXa. En pacientes con Hemofilia, la formación del coágulo se retrasa debido a una notable disminución de la generación de trombina. El coágulo que se forma es inefectivo, produciendose la hemorragia. Como las deficiencias de FVIII y FIX incapacitan la actividad del FX, ostentan cuadros clinicos similares.CLINICA DE LA HEMOFILIA A:
Es importante recordar que la síntesis del factor VIII, se realiza fundamentalmente en las celulas endoteliales de los sinusoides hepáticos y en menor cuantia en otros órganos. La hemostasia es normal, con niveles del factor VIII, superiores al 30%. Los niveles de FVIII se mantienen constantes a lo largo de la vida del paciente y son similares entre los miembros de una misma familia, aunque varian entre familias distintas.
Hemartrosis. (Hemorragias intraarticulares). Constituyen el 75% de los episodios hemorrágicos en pacientes con HA, severa. Lugares de ocurrencia frecuente, son : rodillas, codos, tobillos, hombros, muñecas, caderas. Las hemorragias se anuncian con dolor, continuadas con inflamación, incremento de la temperatura articular y limitación de los movimientos. Si hay fiebre persistente sospechar infección. El sangrado recidivante en una articulación produce hipertrofia sinovial e inflamación con limitación del movimiento y tendencia a más sangrado en esa articulación, causando destrucción del cartílago articular, hiperplasia sinovial y deformidad articular con atrofia muscular y contractura de tejidos blandos.
Hematomas : Generalmente sangrado en musculos o tejidos subcutáneos, hematomas intramusculares, en muslos, glúteos, pantorrillas y antebrazos. Se pueden reabsorber, pero tambien aumentar y causar comprensión de organos, nervios, vasos sanguineos, vias aéreas grandes, hematomas encapsulados (pseudotumores organizados, óseos de Volkmann), que comprimen estructuras vecinas. La principal causa de muerte sigue siendo una hemorragia en el SNC. Todas las hemofilias severas cursan con hematurias asociadas a cólicos renales. Las extracciones dentarias, pueden condicionar hemorragias prolongadas.
{kind=link}

Laboratorio
Alargamiento del TTPa, que se corrige añadiéndole un volumen de plasma normal. El TP y el fibrinógeno : en rango normal. Es necesario cuantificar el Factor para un diagnóstico definitivo. Si simultaneamente se constata un alargamiento del TP, hay que plantear otros diagnósticos o, déficits combinados.
Portadores y diagnóstico prenatal.
El nivel promedio de F:VIII, de una mujer portadora es 50%, aunque algunas pueden cursar con hasta 30%, pudiendo sangrar. El diagnóstico prenatal se realiza en celulas fetales o mediante biopsia de vellosidades corionicas
Diagnóstico, diferencial
Con otras coagulopatias que alargan el TTPa, como déficits de FXI y F:XII, enfermedad de Von Willebrand, inhibidores adquiridos, déficit combinado : FVIII-V .
Tratamiento.
Evitar aspirina e inyecciones IM, tratamiento precóz de sangrados. Tratamiento domiciliario. Planear con cuidado procedimientos quirúrgicos.
Desmopresina (DDAVP), util en hemofilias leves, moderadas y en portadoras sintomáticas a razón de : 0,3 mg/kg/IV, con lo que se incrementa 3 veces la concentración del factor en la mayoria de casos, lográndose un efecto máximo a los 30 minutos.
Concentrados de factor VIII, obtenidos de plasmas humanos, aunque sin inactivar al virus de la hepatitis A y parvovirus. Mas seguros son los concentrados recombinantes.
Dosis de FVIII:
Caso: adulto de 70 kg, con hemorragia en el SNC. Necesita 100% de Factor. Formula: 70x 100/2=3500 UFVIII (dosis de carga o ataque) -Modernamente se prefiere cálculos basados en U/Kg/Peso-. Vida media del FVIII 8-12 horas. Luego de la dosis de ataque se continúa con la mitad de la dosis de carga, c/8 horas
Antifibrinoliticos
Son tratamiento adyuvantes, útiles en extracciones dentales. Contraindicadas si existe hematuria. EACA, oral 5 g (dosis de carga) y luego: 1 g cada 6 hs, durante 8 dias. Acido tranexámico: 1g cada 6 horas. Tapones de fibrina para procedimientos locales, circuncision, extracciones dentales. Los cortes superficiales, se manejan con compresión local. Las epistaxis, las hematurias, profilaxis de endoscopias ameritan 50% de cc plasmática del factor. El sangrado crónico en una articulación, sangrados retrofaringeos o en el SNC:100 %, de cc plasmática del F:VIII.
Profilaxis.
Algunos aconsejan profilaxis con 50 U FVIII/kg/peso, 3 veces a las semana. Se ensaya actualmente terapias génicas y transplante de higado. La profilaxis permanente con factor antihemofilico esté indicada en pacientes con Hemofilia grave, en periodos de crecimiento rápido o mientras estan sometidos a terapia fisica rehabilitadora. En estos casos, se intenta mantener al paciente en valores de Hemofilia leve o moderada.
Evolución y pronóstico
Sin tratamiento adecuado las complicaciones de las hemorragias son recurrentes. Los concentrados de FVIII, como terapia aparecieron en 1960, disminuyendo la morbimortalidad por sangrado. No obstante introdujeron la infección por VIH, la hepatitis B, C e inhibidores adquiridos al FVIII. Desde 1985 se lograron eliminaciones eficaces del virus del VIH y de las Hepatitis de los concentrados.
INHIBIDORES DE FVIII, EN HEMOFILIA A
Son anticuerpos (IgG), variedad IgG4, que interfieren con interacciones del FVIII, con cofactores y activadores.
 Existen los denominados “altos respondedores” con niveles basales mayores de 10 U Bethesda y los “bajos respondedores”, con menos de 10 UB. Ambos se tratan altas dosis de concentrados de FVIII humano o porcino Cuando estas medidas no afectan a los altos respondedores, se les trata con agentes que bypasean al inhibidor. Los “bajos respondedores”, se tratan con FVIII porcino o humano y bypaseadores (FVIIa recombinante). Se arguye que el F:VII activa al FX y que el FXa interactua con Va, convirtiendo la protrombina en trombina.
Existen los denominados “altos respondedores” con niveles basales mayores de 10 U Bethesda y los “bajos respondedores”, con menos de 10 UB. Ambos se tratan altas dosis de concentrados de FVIII humano o porcino Cuando estas medidas no afectan a los altos respondedores, se les trata con agentes que bypasean al inhibidor. Los “bajos respondedores”, se tratan con FVIII porcino o humano y bypaseadores (FVIIa recombinante). Se arguye que el F:VII activa al FX y que el FXa interactua con Va, convirtiendo la protrombina en trombina.
INHIBIDORES ESPONTANEOS, DEL F:VIII.
Presencia de autoanticuerpos en pacientes sin Hemofilia congénita. Aparece en adultos de edad avanzada, en el post-parto de gestantes, en afectas de LES, artritis reumatoide. Responden a métodos de tolerancia inmunológica, prednisona, Ciclofosfamida, Gammaglobulina IV.
Clasificacion Clinica de la Hemofilia A.
Grave o severa: Menos del 1%. Hemorragias espontáneas desde la infancia. Hemartrosis espontáneas
Moderada 1-5%. Hemorragia por trauma o cirugía. Hemartrosis ocasionales
Leve :6-30%. Hemorragia secundaria a trauma o cirugía. Hemorragias espontáneas raras.
Patrones de herencia

-ENFERMEDAD DE VON WILLEBRAND.
Síndrome hereditario autosómico que engloba un grupo bastante heterogéneo de alteraciones cuali-cuantitivas del factor von Willebrand (FvW, cofactor de la ristocetina), una proteina plasmática que trasporta al FVIII y de adhesión de entre las plaquetas y paredes vasculares dañadas, condicionando casi siempre siempre una disminución asociada del FVIII, coagulante. Se sintetiza en celulas endoteliales y megacariocitos. Se almacena en las plaquetas y en los cuerpos de Weibel-Palade, en celulas endoteliales. Su secreción se regula por una serie de multimeros de alto peso molecular. Los de mayor actividad se liberan en respuesta a agentes como la trombina in vitro y el DDAVP, in vivo
TERMINOLOGIADEL FACTOR VON WILLEBRAND Y FACTOR VIII:
-Factor VIII (proteina disminuida en el plasma), de pacientes con HA y EvW.
-Actividad del FVIII: C (propiedad coagulante de la proteina factor VIII).
-Factor VIII antigeno (VIII:Ag), determinante antigenico del FVIII.
-Factor von Willebrand (FvW), gran glucoproteina multimérica necesaria para la adherencia plaquetaria normal, T de Sangria normal y estabilización del FVIII.
Antigeno del factor von Willebrand (FvW:Ag), determinante antigénico del -FvW, cuantificado por métodos inmunológicos.
-Actividad del cofactor de la ristocetina, propiedad del FvW, que produce aglutinación de plaquetas normales.
El FvW desempeña un papel importante en la agregación plaquetaria en los lugares de lesion vascular. Estabiliza el factor VIII, a traves de la formación de enlaces no covalentes entre las 2 proteinas. Se ha descubierto un gran numero de mutaciones del gen del FvW y se han descrito mas de 20 subtipos diferentes de EvW. ooooo oooooo
CLINICA:
Tipo 1:
Constituye el 70 %, de los casos. Falla la molécula antigenica del FvW y la del cofactor de la ristocetina. Es un déficit cuantitativo. Se trasmite con herencia autosomica dominante, de expresión variable y penetrancia incompleta. Los síntomas varian en las familias. Los síntomas puden variar en un paciente a lo largo del tiempo. Los pacientes presentan: epistaxis, equimosis, hematomas, menorragia, sangrado gingival, hemorragia gastrointestinal. Son raras las hemartrosis. Los pacientes con enfermedad leve a moderada, mejoran espontáneamente entre los 20-30 años..Durante el embarazo las afectas mejoran.
Tipo 2
En la variante IIA, faltan los multimeros de peso molecular intermedio y alto.No se detecta el cofactor de la ristocetina.En la variante IIB, aunque faltan los multimeros de peso molecular alto,estos estan presentes en la membrana plaquetaria,por lo que la aglutinación frente a la ristocetina esta conservada. Estos casos cursan con trmbocitopenia.Las variantes IIA y IIB son los trastornos cualitativos mas frecuentes del FvW. Su frecuencia es del orden del 20 30% de los casos.
Tipo 3
Hay carencia absoluta de FvW. Puede existir déficit completo de FVIII:C, pero habitualmente cursan mas bien con valores moderados (3-7%). Es autosomica recesiva. Las que la padecen son homocigotos.
Evolución y pronóstico
Sin tratamiento adecuado las complicaciones de las hemorragias son recurrentes. Los concentrados de FVIII, como terapia aparecieron en 1960, disminuyendo la morbimortalidad por sangrado. No obstante introdujeron la infección por VIH, la hepatitis B, C e inhibidores adquiridos al FVIII. Desde 1985 se lograron eliminaciones eficaces del virus del VIH y de las Hepatitis de los concentrados.
INHIBIDORES DE FVIII, EN HEMOFILIA A
Son anticuerpos (IgG), variedad IgG4, que interfieren con interacciones del FVIII, con cofactores y activadores.
 Existen los denominados “altos respondedores” con niveles basales mayores de 10 U Bethesda y los “bajos respondedores”, con menos de 10 UB. Ambos se tratan altas dosis de concentrados de FVIII humano o porcino Cuando estas medidas no afectan a los altos respondedores, se les trata con agentes que bypasean al inhibidor. Los “bajos respondedores”, se tratan con FVIII porcino o humano y bypaseadores (FVIIa recombinante). Se arguye que el F:VII activa al FX y que el FXa interactua con Va, convirtiendo la protrombina en trombina.
Existen los denominados “altos respondedores” con niveles basales mayores de 10 U Bethesda y los “bajos respondedores”, con menos de 10 UB. Ambos se tratan altas dosis de concentrados de FVIII humano o porcino Cuando estas medidas no afectan a los altos respondedores, se les trata con agentes que bypasean al inhibidor. Los “bajos respondedores”, se tratan con FVIII porcino o humano y bypaseadores (FVIIa recombinante). Se arguye que el F:VII activa al FX y que el FXa interactua con Va, convirtiendo la protrombina en trombina.INHIBIDORES ESPONTANEOS, DEL F:VIII.
Presencia de autoanticuerpos en pacientes sin Hemofilia congénita. Aparece en adultos de edad avanzada, en el post-parto de gestantes, en afectas de LES, artritis reumatoide. Responden a métodos de tolerancia inmunológica, prednisona, Ciclofosfamida, Gammaglobulina IV.
Clasificacion Clinica de la Hemofilia A.
Grave o severa: Menos del 1%. Hemorragias espontáneas desde la infancia. Hemartrosis espontáneas
Moderada 1-5%. Hemorragia por trauma o cirugía. Hemartrosis ocasionales
Leve :6-30%. Hemorragia secundaria a trauma o cirugía. Hemorragias espontáneas raras.
Patrones de herencia
-ENFERMEDAD DE VON WILLEBRAND.
Síndrome hereditario autosómico que engloba un grupo bastante heterogéneo de alteraciones cuali-cuantitivas del factor von Willebrand (FvW, cofactor de la ristocetina), una proteina plasmática que trasporta al FVIII y de adhesión de entre las plaquetas y paredes vasculares dañadas, condicionando casi siempre siempre una disminución asociada del FVIII, coagulante. Se sintetiza en celulas endoteliales y megacariocitos. Se almacena en las plaquetas y en los cuerpos de Weibel-Palade, en celulas endoteliales. Su secreción se regula por una serie de multimeros de alto peso molecular. Los de mayor actividad se liberan en respuesta a agentes como la trombina in vitro y el DDAVP, in vivo
TERMINOLOGIADEL FACTOR VON WILLEBRAND Y FACTOR VIII:
-Factor VIII (proteina disminuida en el plasma), de pacientes con HA y EvW.
-Actividad del FVIII: C (propiedad coagulante de la proteina factor VIII).
-Factor VIII antigeno (VIII:Ag), determinante antigenico del FVIII.
-Factor von Willebrand (FvW), gran glucoproteina multimérica necesaria para la adherencia plaquetaria normal, T de Sangria normal y estabilización del FVIII.
Antigeno del factor von Willebrand (FvW:Ag), determinante antigénico del -FvW, cuantificado por métodos inmunológicos.
-Actividad del cofactor de la ristocetina, propiedad del FvW, que produce aglutinación de plaquetas normales.
El FvW desempeña un papel importante en la agregación plaquetaria en los lugares de lesion vascular. Estabiliza el factor VIII, a traves de la formación de enlaces no covalentes entre las 2 proteinas. Se ha descubierto un gran numero de mutaciones del gen del FvW y se han descrito mas de 20 subtipos diferentes de EvW. ooooo oooooo
CLINICA:
Tipo 1:
Constituye el 70 %, de los casos. Falla la molécula antigenica del FvW y la del cofactor de la ristocetina. Es un déficit cuantitativo. Se trasmite con herencia autosomica dominante, de expresión variable y penetrancia incompleta. Los síntomas varian en las familias. Los síntomas puden variar en un paciente a lo largo del tiempo. Los pacientes presentan: epistaxis, equimosis, hematomas, menorragia, sangrado gingival, hemorragia gastrointestinal. Son raras las hemartrosis. Los pacientes con enfermedad leve a moderada, mejoran espontáneamente entre los 20-30 años..Durante el embarazo las afectas mejoran.
Tipo 2
En la variante IIA, faltan los multimeros de peso molecular intermedio y alto.No se detecta el cofactor de la ristocetina.En la variante IIB, aunque faltan los multimeros de peso molecular alto,estos estan presentes en la membrana plaquetaria,por lo que la aglutinación frente a la ristocetina esta conservada. Estos casos cursan con trmbocitopenia.Las variantes IIA y IIB son los trastornos cualitativos mas frecuentes del FvW. Su frecuencia es del orden del 20 30% de los casos.
Tipo 3
Hay carencia absoluta de FvW. Puede existir déficit completo de FVIII:C, pero habitualmente cursan mas bien con valores moderados (3-7%). Es autosomica recesiva. Las que la padecen son homocigotos.
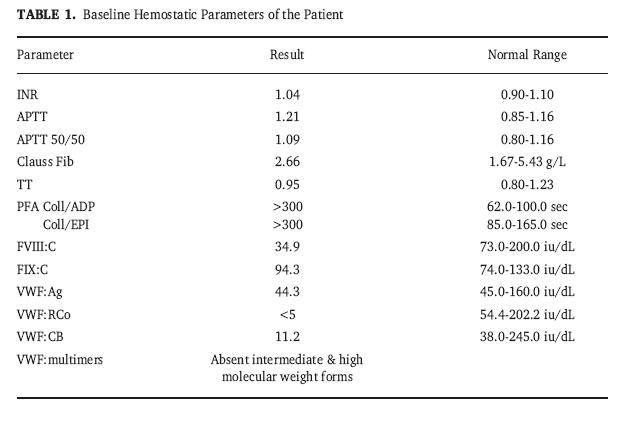 Laboratorio
LaboratorioEn general los parámetros diagnósticos de laboratorio son: 1) Tiempo de Sangria prolongado (N:1-6 minutos.Metodo de Ivy modificado) 2) Disminución de la adhesividad plaquetaria al subendotelio y al cristal 3) Agregación inducida por ristocetina ausente o disminuida 4) FVIII:C, disminuido 5) Factor Von Willebrand, antigenico y cofactor de la ristocetina disminuidos. Por ello, es importante determinar actividad del FVIII, antigeno FvW, actividad del cofactor de la ristocetina, T. de sangria, agregación plaquetaria y analisis de multimeros del FvW. En un mismo sujeto se encuentran variaciones muy amplias de los resultados por lo que se recomiendan estudios repetidos
Diagnostico diferencial.
Con HA, HB y seudo Von Willebrand, entidad similar a la variante IIB, pero inducida por alteración de la membrana plaquetaria y no por multimeros de alto peso molecular..
Enfermedad de von Willebrand adquirida.
Aparece en adultos mayores sin antecedentes personales ni familiares de diatesis hemorrágica. Generalmente está asociada a síndrome mieloproliferativo, linfoproliferativo, tumor solido o defecto cardiaco, empleo de cirprofloxacina, acido valproico o por autoanticuerpos contra FvW
Tratamiento
El objetivo del tratamiento es corregir la deficiencia de FVIII y acortar o corregir el T. de Sangria
DDAVP (Desmopresina)
Los pacientes con EvW tipo I, liberan a la circulación multimeros de un peso molecular alto, 1-3 horas después de la infusión de DDAVP.
 Incrementa los niveles basales de actividad de FVIII, del antigeno FvW, del cofactor de la ristocetina y corrige el T. de Sangria. El 80 %, de los pacientes tipo I, tienen respuestas excelentes al DDAVP. No obstante algunos del tipo II y la totalidad de tipo III no responden al DDAVP. El DDAVP, se emplea en hemorragias leves a moderadas, como profilaxis previa a la cirugía. Una hora antes de la cirugía y después cada 12 horas. Si no es efectivo, se emplean concentrados de FvW y crioprecipitados.
Incrementa los niveles basales de actividad de FVIII, del antigeno FvW, del cofactor de la ristocetina y corrige el T. de Sangria. El 80 %, de los pacientes tipo I, tienen respuestas excelentes al DDAVP. No obstante algunos del tipo II y la totalidad de tipo III no responden al DDAVP. El DDAVP, se emplea en hemorragias leves a moderadas, como profilaxis previa a la cirugía. Una hora antes de la cirugía y después cada 12 horas. Si no es efectivo, se emplean concentrados de FvW y crioprecipitados.
Reposicion de F VWillebrand
Los pacientes que no responden al DDAVP, pueden recibir tratamiento con concentrados de FVIII, que contiene FvW, como el Humate P. tambien: plasma helado fresco, estrógenos y anticonceptivos orales, EACA en forma profiláctica, en procedimientos dentales, en menorragias, epistaxis recurrente. La ventaja de los crioprecipitados frente a los preparados comérciales es su abundancia en multimeros mayores. Los pacientes con EvW, pueden desarrollar autoanticuerpos contra el FvW.
Diagnostico diferencial.
Con HA, HB y seudo Von Willebrand, entidad similar a la variante IIB, pero inducida por alteración de la membrana plaquetaria y no por multimeros de alto peso molecular..
Enfermedad de von Willebrand adquirida.
Aparece en adultos mayores sin antecedentes personales ni familiares de diatesis hemorrágica. Generalmente está asociada a síndrome mieloproliferativo, linfoproliferativo, tumor solido o defecto cardiaco, empleo de cirprofloxacina, acido valproico o por autoanticuerpos contra FvW
Tratamiento
El objetivo del tratamiento es corregir la deficiencia de FVIII y acortar o corregir el T. de Sangria
DDAVP (Desmopresina)
Los pacientes con EvW tipo I, liberan a la circulación multimeros de un peso molecular alto, 1-3 horas después de la infusión de DDAVP.

 Incrementa los niveles basales de actividad de FVIII, del antigeno FvW, del cofactor de la ristocetina y corrige el T. de Sangria. El 80 %, de los pacientes tipo I, tienen respuestas excelentes al DDAVP. No obstante algunos del tipo II y la totalidad de tipo III no responden al DDAVP. El DDAVP, se emplea en hemorragias leves a moderadas, como profilaxis previa a la cirugía. Una hora antes de la cirugía y después cada 12 horas. Si no es efectivo, se emplean concentrados de FvW y crioprecipitados.
Incrementa los niveles basales de actividad de FVIII, del antigeno FvW, del cofactor de la ristocetina y corrige el T. de Sangria. El 80 %, de los pacientes tipo I, tienen respuestas excelentes al DDAVP. No obstante algunos del tipo II y la totalidad de tipo III no responden al DDAVP. El DDAVP, se emplea en hemorragias leves a moderadas, como profilaxis previa a la cirugía. Una hora antes de la cirugía y después cada 12 horas. Si no es efectivo, se emplean concentrados de FvW y crioprecipitados.Reposicion de F VWillebrand
Los pacientes que no responden al DDAVP, pueden recibir tratamiento con concentrados de FVIII, que contiene FvW, como el Humate P. tambien: plasma helado fresco, estrógenos y anticonceptivos orales, EACA en forma profiláctica, en procedimientos dentales, en menorragias, epistaxis recurrente. La ventaja de los crioprecipitados frente a los preparados comérciales es su abundancia en multimeros mayores. Los pacientes con EvW, pueden desarrollar autoanticuerpos contra el FvW.
Labels: Enf.de Von Willebrand, Hemofilias
posted by Victor Mechán Mendez @ 6:24 AM
![]()
0 Comments:
Post a Comment
<< Home