.+Photo+Author.jpg)


PÚPURA TROMBOCITOPÉNICA PROBABLEMENTE INMUNE
UNIVERSIDAD NACIONAL MAYOR DE SAN MARCOS
UNIVERSIDAD NACIONAL MAYOR DE SAN MARCOS
HOSPITAL NACIONAL 2 DE MAYO
Caso: Varón de 23 Años de Edad con Petequias y Equímosis Múltiples en Piel y Mucosa
Alumnos: Mendoza Contreras, Doris; Maximiliano Nieva, Hugo
PRESENTACIÓN DEL CASO
Paciente N.C.C., HC: 1814502, varón de 23 años de edad procedente de Lima que hace 3 días máculas eritematosas en antebrazos, piernas, tobillos, pies y escasas en tórax anterior abdomen y región lumbar bilateral que incrementan de diámetro hasta 0,5cm aproximadamente.
Con esta sintomatología el día 29.10.06 acude a Clínica San Juan Bautista en San Juan de Lurigancho, donde indican que visite un especialista del Hospital Nacional Dos de Mayo para descartar una posible púrpura. Allí, se solicita un recuento de plaquetas, cuyo resultado es 10 000xmm3.
Al día siguiente (30.10.06) nota mancha eritémato-violácea en el lado izquierdo de la lengua que llega a medir 2cm de diámetro (Fig.5), que ocasiona dolor a la ingestión, por todo esto acude al servicio de Hematología de este nosocomio, donde indican su hospitalización.
FUNCIONES BIOLÓGICAS
Sin alteración.
ANTECEDENTES
PERSONALES
Generales
-Ocupaciones anteriores: construcción civil, gasfitería, en contacto con cementos y pegamentos, refiere que este último ocasiona esporádicamente tos, dolor de ”garganta”.
Fisiológicos:
-Nacido de parto eutócico.
-Desarrollo psicomotor normal.
-Desarrollo psicomotor normal.
Patológicos:
-A los 8 años Cólera, hospitalizado durante dos meses.
-Hace aproximadamente 1 año presenta cuadro similar de igual localización, también en amígdalas, de menor tamaño, acompañado de gingivorragia y cansancio, por lo que acude a farmacia donde le indican ampolla y 2 pastillas diarias por 7 días cuyo nombre no recuerda, con lo cual remiten las lesiones.
FAMILIARES
-madre: migraña.
-hermana (21 años): alergia al polvo.
-hermana (18 años): masa tumoral en seno.
EXAMEN FISICO:
Funciones Vitales:
* PA: 110/70 mmHg
* F de pulso:90 x min.
* FR: 20 x min
* Temperatura: 36.5 ºC
Paciente en buen estado general, en buen estado de hidratación y buen estado de nutrición. Piel tibia, consistente, presenta máculas y pápulas eritematovioláceas de 1-5mm de diámetro en lado izquierdo de cuello (Fig.1), tórax anterior, piernas (Fig. 3 y 4), pies, escasas en región dorso lumbar y cuero cabelludo, que no desaparecen a la vitropresión. Equímosis en labio superior de 5mm de diámetro. Mucosas hidratadas, no pálidas, lesión úlcero-costrosa sobre equímosis de 2cm de diámetro en borde izquierdo de lengua. Uñas: palidez , cianosis distal leve, onicodistrofia en primer ortejo izquierdo, llenado capilar <2seg.
* Temperatura: 36.5 ºC
Paciente en buen estado general, en buen estado de hidratación y buen estado de nutrición. Piel tibia, consistente, presenta máculas y pápulas eritematovioláceas de 1-5mm de diámetro en lado izquierdo de cuello (Fig.1), tórax anterior, piernas (Fig. 3 y 4), pies, escasas en región dorso lumbar y cuero cabelludo, que no desaparecen a la vitropresión. Equímosis en labio superior de 5mm de diámetro. Mucosas hidratadas, no pálidas, lesión úlcero-costrosa sobre equímosis de 2cm de diámetro en borde izquierdo de lengua. Uñas: palidez , cianosis distal leve, onicodistrofia en primer ortejo izquierdo, llenado capilar <2seg.
Tejido celular subcutáneo conservado. Sistema linfático sin alteraciones. Aparato locomotor sin alteraciones. Sistema osteoarticular sin alteraciones.Aparato Respiratorio y Cardiovascular sin alteraciones.
Abdomen:
Simétrico, móvil con la respiración; ruidos hidroaéreos presentes, no dolor a la palpación superficial y profunda, se palpa hígado a 2 cm DRCD, bazo no se palpa, ni se percute.
Sistema génito-urinario, neurológico sin alteraciones
Sistema vascular periférico:
Pulsos periféricos de intensidad incrementada, simétricos, vasos colapsables.
Sistema vascular periférico:
Pulsos periféricos de intensidad incrementada, simétricos, vasos colapsables.
DIAGNÓSTICO CLÍNICO
Síndrome hemorrágico.
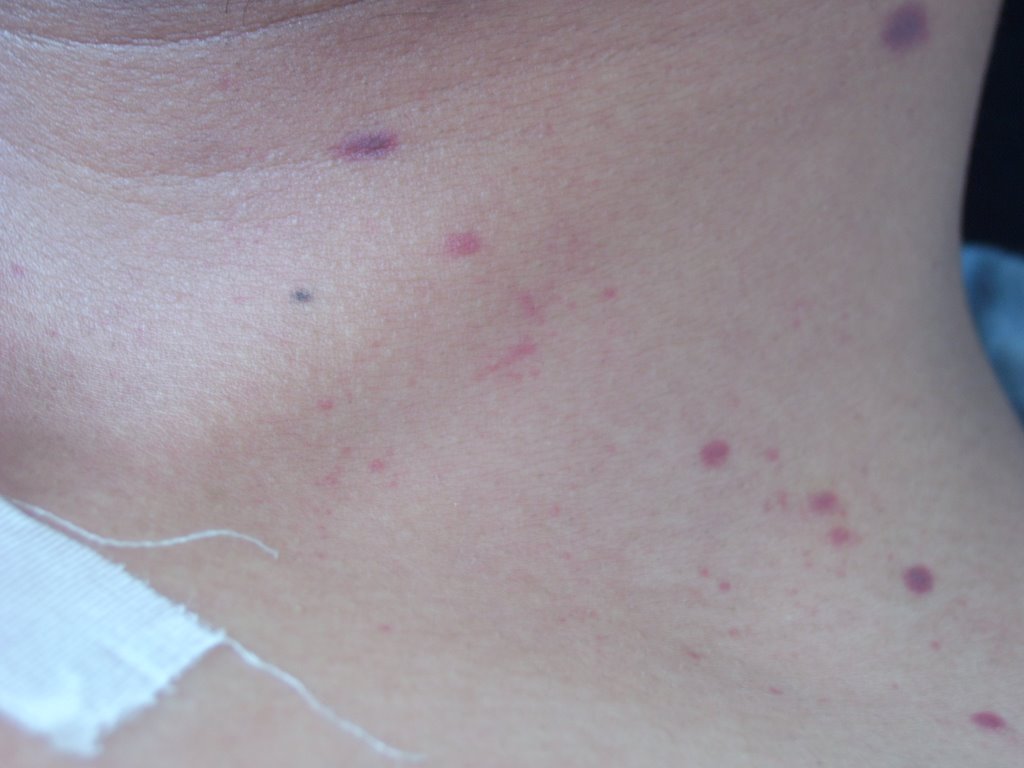 Fig.1: Petequias en lado derecho de cuello (31.10.06)
Fig.1: Petequias en lado derecho de cuello (31.10.06) 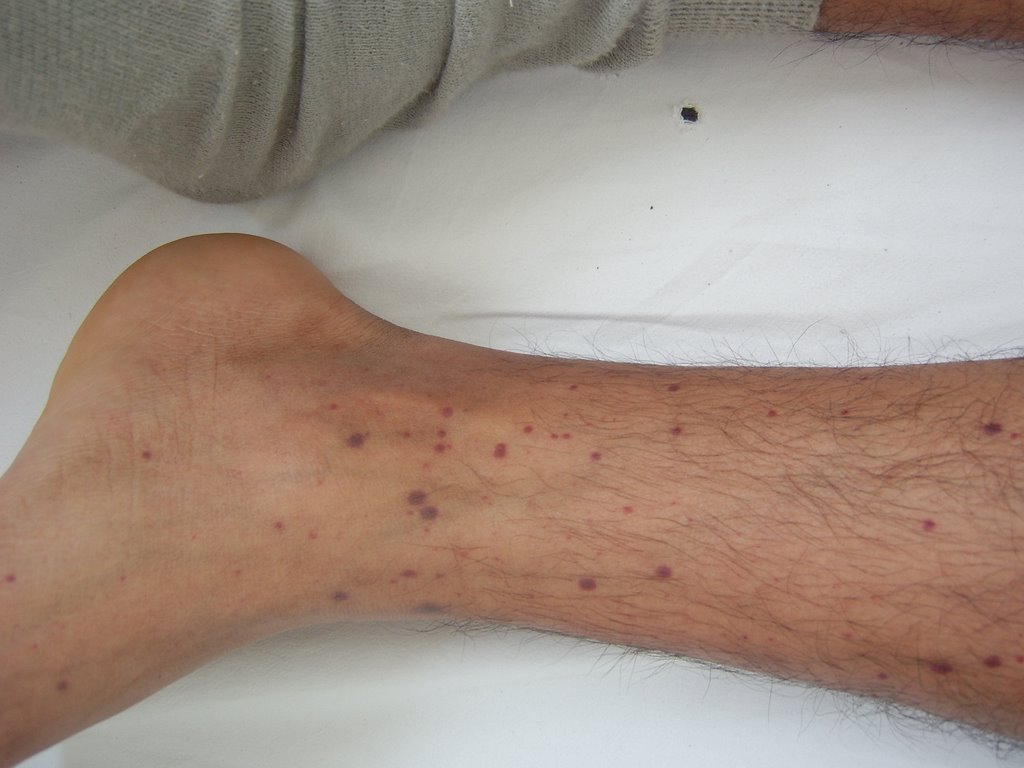 Fig.2: Petequias en pierna y pie izquierdo (31.10.06)
Fig.2: Petequias en pierna y pie izquierdo (31.10.06)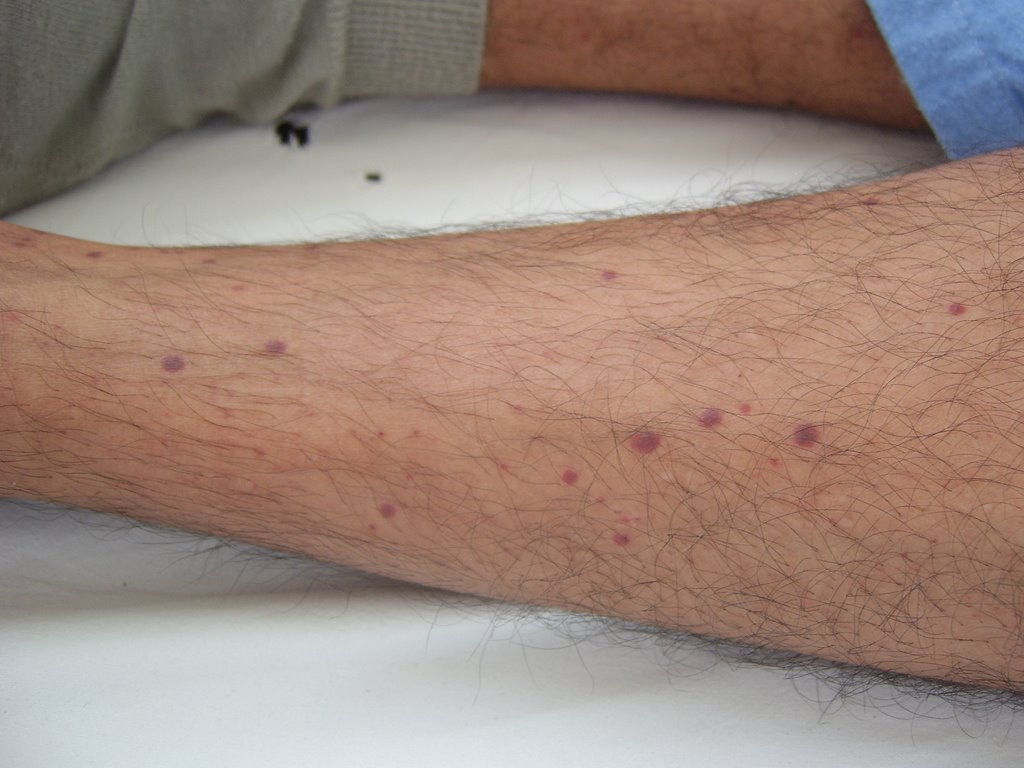
Fig.3: Petequias en región pretibial izquierda (31.10.06)
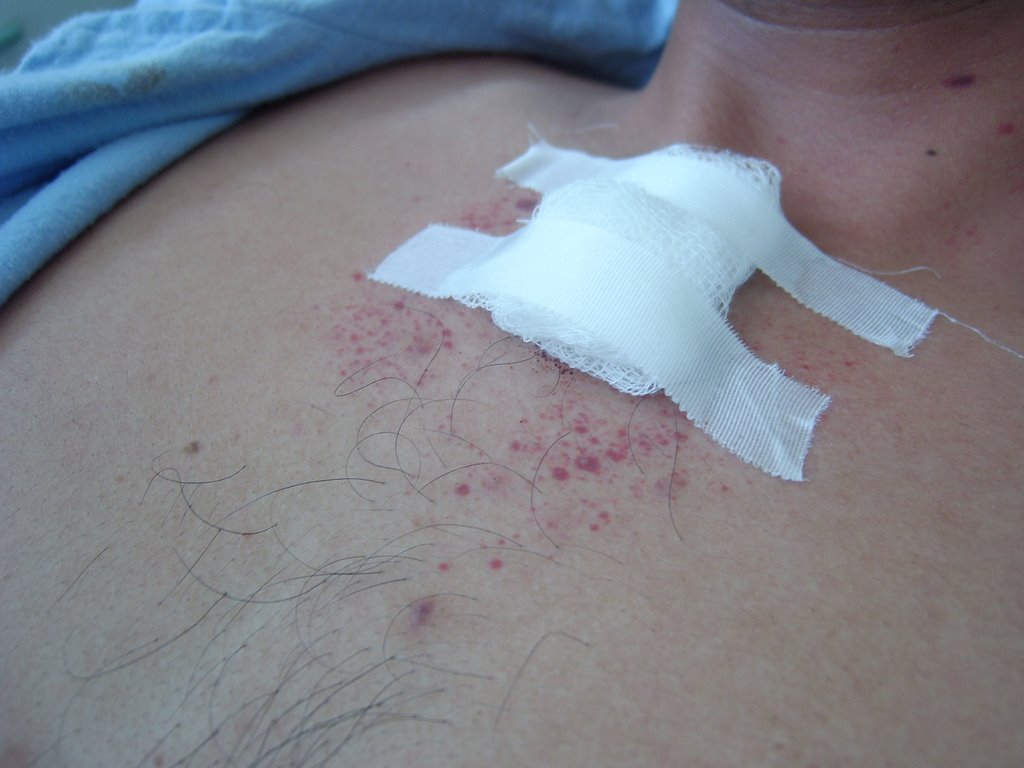 Fig.4: Petequias en región precordial, luego de
Fig.4: Petequias en región precordial, luego de punción para aspirado medular (31.10.06)
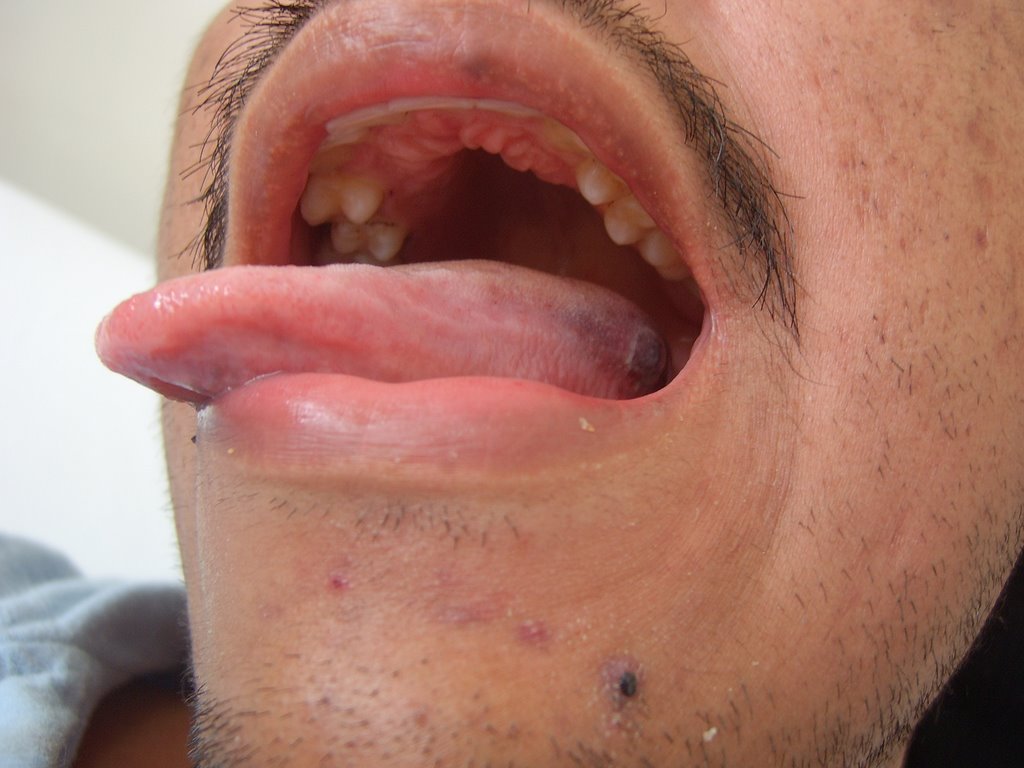 Fig.5: Equímosis en labio superior, lesión úlcera costrosa sobre
Fig.5: Equímosis en labio superior, lesión úlcera costrosa sobre equímosis en borde izquierdo de lengua (31.110.06)
EXÁMENES AUXILIARES






Aspirado de Médula Ósea: 30.10.06
Conclusión:
1. Ausencia de hemosiderina
2. Hiperplasia megacariocítica
3. Incremento del tejido adiposo
2. Hiperplasia megacariocítica
3. Incremento del tejido adiposo
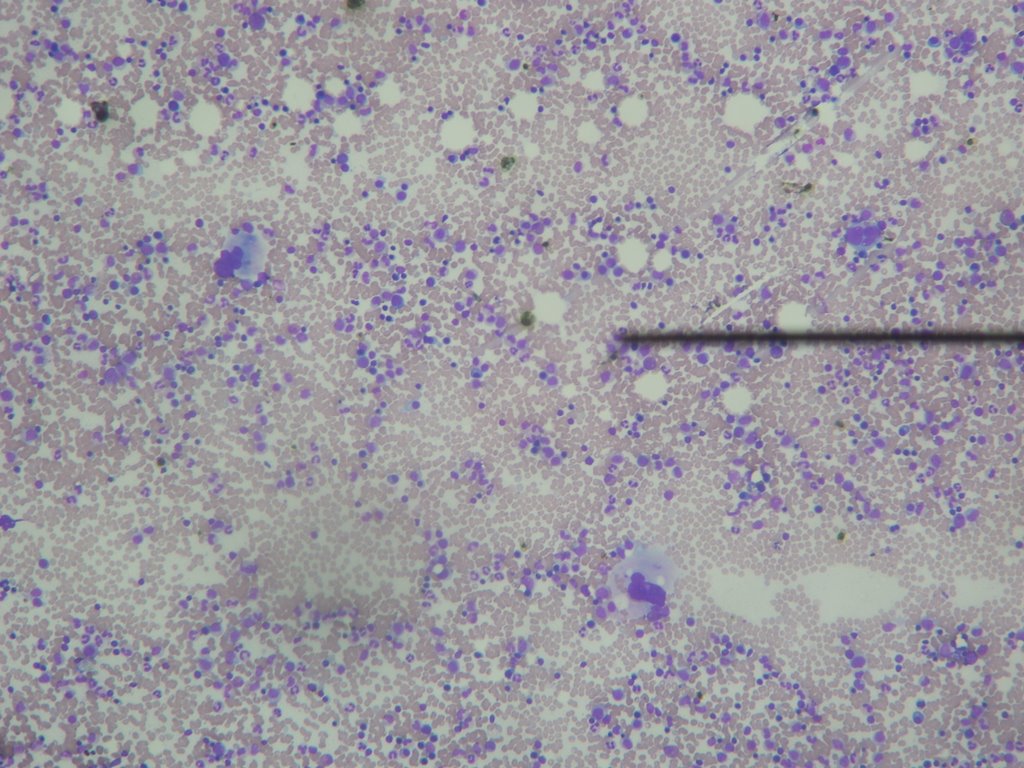 Fig. 6: Megacariocitos en Aspirado Medular (31.10.06)
Fig. 6: Megacariocitos en Aspirado Medular (31.10.06)
TRATAMIENTO
30.10.06
Dexametasona 8 mg 1 amp EV c/8h
Ranitidina 50 mg EV c/8h
No Intramusculares
Luego de tres días, la dexametasona fue sustituida: por Prednisona 75 mg VO c/24h y la ranitidina a 30 mg VO a 8:00pm.
El paciente fue dado de alta el 04.11.06, con las indicaciones señaladas.
DISCUSIÓN
El síndrome hemorrágico puede deberse a trastornos vasculares, plaquetarios o de la coagulación. La objetivación de una lesión de morfología máculo-pápular, de color rojizo que no desaparece a la vitropresión, nos conlleva a la observación de la morfología de la lesión, si la lesión es macular, púrpura no palpable orientará a etiología no vascular; si la lesión es papular, púrpura palpable, la patología vascular: por tanto nuestras lesiones no se encuadran en este algoritmo.
El síndrome purpúrico comprende el grupo de enfermedades en las que se producen pequeñas hemorragias de las capas superficiales de la piel o mucosas dando una coloración purpúrea, (falla de la hemostasia primaria). La trombocitopenia que se presenta limita nuestro panorama a tres posibles causas, entre ellas: producción defectuosa, aumento de la destrucción y secuestro plaquetario. Una producción defectuosa se asocia a plaquetas de reducido tamaño, las cuales no se observan en los hemogramas; por otro lado en el secuestro plaquetario se evidencia hiperesplenismo, que no ha sido hallazgo del examen físico, por tanto correspondería a un incremento de la destrucción plaquetaria.
El abordaje debe orientarse a dos objetivos: apreciar el riesgo hemorrágico e investigar la causa con la ayuda de un mielograma, después de haber eliminado otras causas evidentes. Este último permite determinar el origen central o periférico de la trombopenia, en este caso se trata del tipo periférico.
Dentro de las púrpuras trombocitopénicas tenemos las de tipo no inmune e inmune. En el primer caso se tiene por ejemplo al síndrome urémico-hemolítico, púrpura trombótica trombocitopénica, hemangiomas, circulación turbulenta, CID, fármacos, entre otras, donde nuestra historia clínica y exámenes auxiliares alejan la posibilidad de alteración neurológica, una anemia hemolítica, insuficiencia renal aguda, entre otros que podrían presentarse en estos casos.
El diagnóstico de una púrpura trombocitopénica inmune se hace por exclusión de formas secundarias, p.e. las asociadas a Lupus eritematoso sistémico, síndrome antifosfolipidico, estados de inmunodeficiencia, desórdenes linfoproliferativos, infección con el virus de inmunodeficiencia humana y el virus de la Hepatitis C, y terapia con drogas como la heparina y quinidina. Es importante tener en cuenta que una trombocitopenia hereditaria puede ser enmascarada como púrpura trombocitopénica inmune. La ausencia de antecedentes familiares, síntomas sistémicos, examen clínico y pruebas de laboratorio nos ayuda a excluir estas formas secundarias.
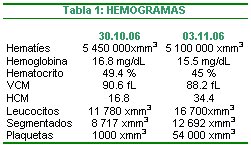
Es necesario 5 criterios diagnósticos: I) trombocitopenia en por lo menos dos recuentos en días diferentes II) exclusión de otras enfermedades III) hiperplasia megacariocítica IV) presencia de anticuerpos antiplaquetarios y V) respuesta al tratamiento autoinmune. Se presenta dos hemogramas en que se observa plaquetopenia (Tab 1), se excluyeron varias patologías mediante historia clínica y examenes de laboratorio, se evididencia la hiperplasia megacariocítica en el aspirado de médula ósea y por último se obtiene respuesta al tratamiento inmune con una variación de 1 000 a 54 000xmm3; se cumple con cuatro de cinco criterios, por lo tanto existe una alta probabilidad de tratarse de una Púrpura Trombocitopénica Inmune de presentación en adultos.
El recuento plaquetario inicial es 1 000xmm3 , la literatura refiere que un recuento de plaquetas inferiores a 20.000 conlleva a mayor riesgo de hemorragias internas y sangrado del sistema nervioso central, hallazgo que clínicamente no se evidenció, debido probablemente a que no hubo persistencia de esta magnitud de trombocitopenia.
Dentro de las púrpuras trombocitopénicas tenemos las de tipo no inmune e inmune. En el primer caso se tiene por ejemplo al síndrome urémico-hemolítico, púrpura trombótica trombocitopénica, hemangiomas, circulación turbulenta, CID, fármacos, entre otras, donde nuestra historia clínica y exámenes auxiliares alejan la posibilidad de alteración neurológica, una anemia hemolítica, insuficiencia renal aguda, entre otros que podrían presentarse en estos casos.
El diagnóstico de una púrpura trombocitopénica inmune se hace por exclusión de formas secundarias, p.e. las asociadas a Lupus eritematoso sistémico, síndrome antifosfolipidico, estados de inmunodeficiencia, desórdenes linfoproliferativos, infección con el virus de inmunodeficiencia humana y el virus de la Hepatitis C, y terapia con drogas como la heparina y quinidina. Es importante tener en cuenta que una trombocitopenia hereditaria puede ser enmascarada como púrpura trombocitopénica inmune. La ausencia de antecedentes familiares, síntomas sistémicos, examen clínico y pruebas de laboratorio nos ayuda a excluir estas formas secundarias.
Es necesario 5 criterios diagnósticos: I) trombocitopenia en por lo menos dos recuentos en días diferentes II) exclusión de otras enfermedades III) hiperplasia megacariocítica IV) presencia de anticuerpos antiplaquetarios y V) respuesta al tratamiento autoinmune. Se presenta dos hemogramas en que se observa plaquetopenia (Tab 1), se excluyeron varias patologías mediante historia clínica y examenes de laboratorio, se evididencia la hiperplasia megacariocítica en el aspirado de médula ósea y por último se obtiene respuesta al tratamiento inmune con una variación de 1 000 a 54 000xmm3; se cumple con cuatro de cinco criterios, por lo tanto existe una alta probabilidad de tratarse de una Púrpura Trombocitopénica Inmune de presentación en adultos.
El recuento plaquetario inicial es 1 000xmm3 , la literatura refiere que un recuento de plaquetas inferiores a 20.000 conlleva a mayor riesgo de hemorragias internas y sangrado del sistema nervioso central, hallazgo que clínicamente no se evidenció, debido probablemente a que no hubo persistencia de esta magnitud de trombocitopenia.
El tratamiento que recibió fue inicialmente Dexametasona 8 mg EV c/8h por 3 días y luego fue sustituida por Prednisona 75 mg VO c/24 horas. Esta última permite que 2/3 de los pacientes eleven sus recuentos plaquetarios por encima de 50 000/uL. Los esteroides mejoran el adelgazamiento del endotelio vascular y eliminan la púrpura antes que se eleven las plaquetas. Pero también es importante tener presente efectos adversos sobre la mucosa gástrica, por ello la indicación de un inhibidor de los receptores H2 de la histmanina. La evaluación a los cinco días de tratamiento fue favorable, se evidenció una variación de 1 000 a 54 000Xmm3, se cumplió con los cuidados especiales durante el internamiento como la no colocación de inyecciones intramusculares o accesos venosos centrales.
El paciente fue dado de alta en 04.11.06 con la medicación indicada.
REFERENCIAS
Covarrubias R, Sotelo N, Hurtado J. Púrpura trombocitopénica autoinmune.Bol Med Hosp Infant Mex 2004; 61(2) : 119-127.
Zimmer J, Andrés E, Noel E, Koumarianou A. Current management of adult idiopathic thrombocytopenic purpura in practice: a cohort study of 201 patients from a single center. Clin. Lab. Haem. 2004, 26, 137–142.
Douglas C, Blanchette V, Chir B. Immune thrombocypenic purpura. N ENGL J MED, Vol. 346, No. 13 March 28, 2002.
Cheng Y, Wong R, Soo Y, Chung Hin Chui, Fung Yi Lau, Chan N, Wai Shan Wong and Cheng G. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N ENGL J MED 349;9 August 28, 2003.
Bah M, Houery M. Actuación ante las anomalías cuantitativas y cualitativas de las plaquetas. Acta Bioquím. Clín. Latinoam. v.39 n.3 La Plata jun./sept. 2005.
Madero L, Molina J, Sevilla J. Púrpura Trombocitopénica Idiopática. Controversias.
BSCP Can Ped 2001; 25- nº 2.
Labels: immune trombocythopenia, PTI
posted by Victor Mechán Mendez @ 11:10 PM
![]()
1 Comments:
necesitoinscribirme para hacer revisiones bibliograficas de salud
Post a Comment
<< Home