.+Photo+Author.jpg)


LINFOMA DE HOGDKINS. http://es.youtube.com/watch?v=U3JDBvzmECM
El linfoma de Hodgkins, es una neoplasia del tejido linfoide, caracterizada histológicamente por la presencia de células de REED STERNBERG (RS), de gran tamaño, citoplasma claro y 2 o, más núcleos de cromatina reticulada, con prominentes nucleolos eosinofilicos, separados por espacios claros a consecuencia del engrosamiento de sus membranas nucleares. Se las considera de origen celular B, con reordenaciones genéticas en sus inmunoglobulinas. Las células RS, representan supercrecimientos monoclonales de los centros germinales de las células B, que han sufrido mutaciones somáticas extensas, en el curso de respuestas inmunes a noxas foráneas. Presentan cariotipo hiperdiploide, con anomalias estructurales, pero sin aberraciones cromosómicas patognomónicas. Las células RS, secretan citoquinas y quimiocinas (TGF-b = estimula proliferación de fibroblastos, IL-5 = factor de crecimiento de eosinófilos, IL-1= induce fiebre y libera reactantes de fase aguda, IL-10 y TGF-b = con efectos inmunosupresores, responsables del reclutamiento (90%), de celulas no cancerigenas, constituyentes de la mayor parte de la población celular del tumor. Una variante de la célula RS, es la CELULA DE HODGKINS, tipicamente mononucleada. Todos los pacientes con LH, presentan anomalias en su inmunidad celular, persistiendo estos defectos incluso después de tratamientos satisfactorios. En la mitad de casos de LH, se demuestra la presencia del virus de Epstein Barr (EB). La etiologia infecciosa es sugerida tambien por la evidencia de que la Mononucleosis Infecciosa (MI), serológicamente confirmada, confiere un riesgo 3 veces superior para adquirir LH, en adultos jóvenes. Existirian factores genéticos de susceptibilidad al LH. Es probable que el LH, sea el resultado de una infección viral adquirida en la infancia, que se manifiesta al cabo de varios años, en quienes concurran factores genéticos y ambientales.
-EPIDEMIOLOGIA.
Su incidencia es :1-3 casos /100 000 habitantes. Predomina ligeramente en varones. Existe un pico de presentación alrededor de los 20 y despues de los 50 años. La incidencia parece estar influenciada por factores socioeconómicos y ambientales.
-CLINICA.
-La EH, se presenta generalmente con adenopatias localizadas en 1 o 2 regiones laterocervicales, con incremento indoloro de ganglios linfáticos. Ganglios palpables : cervicales (60-80%), axilares (9-20%), inguinales (6-12%). Algunos pacientes presentan Signo de Hoster (dolor en adenopatias trás ingesta de bebidas alcohólicas). Existen síntomas B, cuando estan presentes : Fiebre, mayor a 38 C, sudor nocturno y perdida ponderal, superior al 10% del peso basal. Fiebre de Pel-Ebstein, durante 1-2 semanas alternada con periodos afebriles de similar duración. La enfermedad intratorácica (adenopatias mediastinicas y otros), está presente en 2/3 de los pacientes en el momento del diagnóstico.
-EPIDEMIOLOGIA.
Su incidencia es :1-3 casos /100 000 habitantes. Predomina ligeramente en varones. Existe un pico de presentación alrededor de los 20 y despues de los 50 años. La incidencia parece estar influenciada por factores socioeconómicos y ambientales.
-CLINICA.
-La EH, se presenta generalmente con adenopatias localizadas en 1 o 2 regiones laterocervicales, con incremento indoloro de ganglios linfáticos. Ganglios palpables : cervicales (60-80%), axilares (9-20%), inguinales (6-12%). Algunos pacientes presentan Signo de Hoster (dolor en adenopatias trás ingesta de bebidas alcohólicas). Existen síntomas B, cuando estan presentes : Fiebre, mayor a 38 C, sudor nocturno y perdida ponderal, superior al 10% del peso basal. Fiebre de Pel-Ebstein, durante 1-2 semanas alternada con periodos afebriles de similar duración. La enfermedad intratorácica (adenopatias mediastinicas y otros), está presente en 2/3 de los pacientes en el momento del diagnóstico.

 Una minoria, tiene afectación subdiafragmática, exclusivamente. La frecuencia de afectación esplénica en el momento de la laparotomia en pacientes no tratados es :37%. La afectación esplénica depende del tipo histológico, estando presente en el 60 % de los casos de celularidad mixta y depleción linfocitica, en contraste a un 34% de casos con predominio linfocitico o, esclerosis nodular. La enfermedad diseminada es mas común en casos de celularidad mixta y depleción linfocitica. Cuando la presentación es muy tumoral, ocurren trastornos por compresión (Sindrome de vena cava superior, compresión medular o ureterohidronefrosis), infiltración de órganos y tejidos.
Una minoria, tiene afectación subdiafragmática, exclusivamente. La frecuencia de afectación esplénica en el momento de la laparotomia en pacientes no tratados es :37%. La afectación esplénica depende del tipo histológico, estando presente en el 60 % de los casos de celularidad mixta y depleción linfocitica, en contraste a un 34% de casos con predominio linfocitico o, esclerosis nodular. La enfermedad diseminada es mas común en casos de celularidad mixta y depleción linfocitica. Cuando la presentación es muy tumoral, ocurren trastornos por compresión (Sindrome de vena cava superior, compresión medular o ureterohidronefrosis), infiltración de órganos y tejidos.-MAL PRONOSTICO: Explicitado por la presencia de :signo-sintomatologia B, masa tumoral alta (masa mediastinica de diámetro superior al 1/3 torácico), afección de múltiples territorios, más de 5 nódulos de EH en el bazo. VSG elevada, depleción linfocitica, sexo masculino, mas de 45 años, estadio IV, leucocitos: mas de 15 000/mm3, linfocitos menos de 600/mm3, rapidez de respuesta a la terapia, Hb menos de 10, 5 gr/dl, albumina menos de 4 gr/dl, niveles elevados de beta 2-microglobuina y de citoquinas (IL-6, IL-10, CD30, CD25). La presencia de un factor, condiciona una supervivencia reducida en un 7%. Con 5-7 factores la probabilidad de supervivencia libre de progresión, a los 5 años, es: 42%),
-CORRELACION CLINICO-PATOLOGICA: Nódulo linfatico reactivo normal

-LINFOMA RICO EN LINFOCITOS (NODULAR): 
Requiere técnicas inmunohistoquimicas para su diagnóstico. Usualmente, la célula de RS, expresa CD30 y CD15,
pero carece de expresión : CD45 o, CD20. La mayoria de sus componentes, son celulas B normales, pequeñas organizadas en patrón nodular o folicular. Compromete ganglios axilares. Ocasionalmente, algunos linfocitos y celulas de aspecto histiocitario -de estirpe B- carecen de antigenos de activacion como el CD15, lo que unido a su curso indolente ha instado a algunos a separarla del resto de variedades de LH.
-PREDOMINIO LINFOCITICO: Presente en el 10%, de los pacientes. El estadio I, aparece en el 70%, de los casos.
-ESCLEROSIS NODULAR.
En esta variedad

Requiere técnicas inmunohistoquimicas para su diagnóstico. Usualmente, la célula de RS, expresa CD30 y CD15,

pero carece de expresión : CD45 o, CD20. La mayoria de sus componentes, son celulas B normales, pequeñas organizadas en patrón nodular o folicular. Compromete ganglios axilares. Ocasionalmente, algunos linfocitos y celulas de aspecto histiocitario -de estirpe B- carecen de antigenos de activacion como el CD15, lo que unido a su curso indolente ha instado a algunos a separarla del resto de variedades de LH.
-PREDOMINIO LINFOCITICO: Presente en el 10%, de los pacientes. El estadio I, aparece en el 70%, de los casos.
-ESCLEROSIS NODULAR.
En esta variedad

existen amplias bandas de fibrosis y abundantes CELULAS LACUNARES, variantes de células de RS, mononucleadas, 

con halo claro alrededor del citoplasma, debido a retracción por efecto de fijación. Recientemente se ha descrito la variante SINCITIAL, en la que celulas atipicas se disponen en acumulos o, bandas. Presente en el 40-70% de los pacientes. – Se presenta en mujeres jóvenes, afectando los ganglios cervicales, inferiores supraclaviculares y mediastinicos. El 70%, cursa con un estadio limitado de la enfermedad.
-CELULARIDAD MIXTA. Presenta signos B.

Presenta signos B. Es el tipo menos frecuente. Existen muchas celulas pleomorficas junto a celulas de RS y pocos linfocitos. Hay 2 subtipos: a-Reticular :abundantes celulas neoplasicas pleomorficas. b-Fibrosis difusa :subtipo mas comun con proliferación fibroblástica. En adultos, la enfermedad es mas extensa. Frecuentemente dispone de sintomas sistémicos.
-ESTADIAJE:

Para establecer el estadiaje se necesita: Radiografia de tórax. TC torácica y abdominal, biopsia de hueso e higado. En determinados casos laparotomia exploradora con esplenectomia. Hoy dia solo se considera a la laparotomía para el estadiaje de candidatos a radioterapia. Linfangiografia bipedal: la mitad de los pacientes con afectación de ganglios supradiafragmaticos, cursa con afectación infradiafragmática. Este procedimiento esta siendo descartado por engorroso.
-LABORATORIO
Incremento de la VSG, linfopenia. Con menos frecuencia eosinofilia e incremento de la fosfatasa alcalina.-Inmunofenotipicamente, la celula RS, corresponde a linfocitos con marcadores de activacion : CD30 +, CD15 +, CD 25 +, HLA-DR +, si bien el antigeno CD45 (antigeno leucocitario comun) y el CD20 (pan marcador B), son negativos, lo que ayuda a diferenciarlos del LNH. En el 85% de los casos de esclerosis nodular y celularidad mixta (y en una pequeña proporción de los otros subtipos), la célula de RS expresa niveles variables de CD30, aunque carece generalmente de expresión de CD15. Los procedimientos diagnósticos: emisión de positrones (EP), TAC y RM tienen la misma sensibilidad y especificidad, para determinar ganglios crecidos..
-TRATAMIENTO.
-RADIOTERAPIA. Dosis habituales.
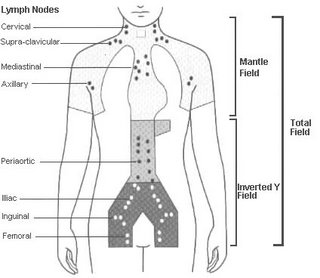
-Campo implicado: de (35 -44 Gy) a (150-200 Gy), por fracción diaria (5 dias x semana)
-Campos no implicados : profiláctica de (30-35 Gy).
Definición de los campos:
Manto : cervical, supraclavicular, infraclavicular, mediastino y ganglios hiliares
Paraaórtico: bazo y pediculo esplénico.
Manto + paraaórtico : irradiación linfoide subtotal
Paraaórtico + pélvico :Y, invertida.
Manto + Y, Invertida : irradiación linfoide total.
-QUIMIOTERAPIA
-MOOP (mecloretamina, vincristina, procarbazina, prednisona)
-44% de los pacientes con enfermedad avanzada, libres de enfermedad, 20 años después
-AVBD (adriamicina, bleomicina, vinblastina y DTIC)
Eficáz en fallos de MOPP. Alternada con MOOP= regimenes alternantes o, hibridos
-REGIMENES ALTERNATIVOS, Empleados en enfermedad avanzada, incorporando G-CSF, para mitigar mielotoxicidad, por altas dosis.
-BEACOPP (bleomicina, etoposido, doxorubicina, ciclofosfamida, vincristina, procarbazina, prednisona
-Stanford V.
-ENFERMEDAD FAVORABLE, ESTADIO LIMITADO
-Estadio :IA/IIA, supradiafragmatica sin masas tumorales: radioterapia (manto o invertida) : 95-100 %, de supervivencia a los 5 años. Otra opción es administrar 3 ciclos de AVBD, continuada con radioterapia de campos afectados.
-ESTADIO LIMITADO, LOCALMENTE AVANZADA
Estadio : IA/IIA, con masa mediastinica voluminosa, (mas de 1/3, del diametro toracico abarcado por el tumor), caracterizada por presentar altas tasas de recaida con radioterapia, necesita de una modalidad de tratamiento combinada (80% de supervivencia libre de enfermedad, en comparación con el 50 %, con radioterapia sola). Para esta afección es mejor emplear combinaciones de radioterapia y quimioterapia. Afectación de ninguno o, solo un sitio extranodal : Irradiación en campo extendido (linfoide subtotal), como tratamiento de elección (80 %, libre de enfermedad a los 15 años). La quimioterapia combinada es curativa en 75-80%, de casos. Aunque el tratamiento del estadio IB/ IIB, sigue siendo controvertido, suele implicar el uso de quimioterapia o una modalidad combinada, dado que se le considera enfermedad diseminada.
-ENFERMEDAD AVANZADA
-La terapia en estadio IIIA, esta en debate. El problema es ¿la radioterapia implica irradiacion ganglionar total o es preferible la quimioterapia sistemica?. -La quimioterapia sistémica tambien esta en debate. ¿MOOP frente a AVBD, MOPP/AVBD o, nuevos regimenes : :BEACOPP, Stanford V. Parece que AVBD es preferible a hibridos, debido a su menor toxicidad. No obstante, el pronóstico mejora empleando: BEACOPP. En la actualidad se trata con radioterapia total a los enfermos con poca masa tumoral y con qumioterapia al resto. Para los estadios : IIIB y IV, se prefiere multiquimioterapia.
-ENFERMEDAD RECURRENTE
Las recidivas locales, se tratan con radioterapia. Para las recaidas sistemicas, si se empleo MOPP y la remision duro 1 año, emplear AVBD (40% de respuestas).-Recaida tras radioterapia: la qumioterapia logra una excelente tasa de curación, empleando 7 farmacos o mas. Si se emplearon terapias hibridas o alternantes -y hay fracaso- lo mejor es recurrir a trasplantes autologos de medula osea (40%, de sobrevida a los 5 años), o de celulas precursoras perifericas.
-COMPLICACIONES
-Neoplasias secundarias: LMA, mielodisplasia (1-10%, en 10 años), dependiente de dosis acumuladas de alquilantes. Riesgo de Linfoma agresivo difuso de células B, cánceres sólidos: pulmón, estómago, hueso, tejidos blandos (18% a los 15 años), dependiendo del grado de exposición a la quimioterapia. Puede desarrollarse una enfermedad cardiaca en receptores de radiación mediastinica. Es frecuente la infertilidad, tras tratamiento con MOPP. Puede aparecer una neumonitis por radiación, dependiendo de la dosis recibida por por el pulmon. Se producen anomalias de funcion tiroidea en cerca del 30% de los pacientes, tras irradiación tipo manto.
-CELULARIDAD MIXTA. Presenta signos B.

Presente en el 30-50% de pacientes. Es común diagnosticar la enfermedad en estadio avanzado, tanto en pacientes pediatricos como en otros de edad avanzada. Se asocia al virus de EB. Presenta abundantes celulas de RS, junto a linfocitos, eosinófilos, células plasmáticas e histiocitos.
-DEPLECION LINFOCITICA.
-DEPLECION LINFOCITICA.
Presenta signos B. Es el tipo menos frecuente. Existen muchas celulas pleomorficas junto a celulas de RS y pocos linfocitos. Hay 2 subtipos: a-Reticular :abundantes celulas neoplasicas pleomorficas. b-Fibrosis difusa :subtipo mas comun con proliferación fibroblástica. En adultos, la enfermedad es mas extensa. Frecuentemente dispone de sintomas sistémicos.
-ESTADIAJE:

Para establecer el estadiaje se necesita: Radiografia de tórax. TC torácica y abdominal, biopsia de hueso e higado. En determinados casos laparotomia exploradora con esplenectomia. Hoy dia solo se considera a la laparotomía para el estadiaje de candidatos a radioterapia. Linfangiografia bipedal: la mitad de los pacientes con afectación de ganglios supradiafragmaticos, cursa con afectación infradiafragmática. Este procedimiento esta siendo descartado por engorroso.
-LABORATORIO
Incremento de la VSG, linfopenia. Con menos frecuencia eosinofilia e incremento de la fosfatasa alcalina.-Inmunofenotipicamente, la celula RS, corresponde a linfocitos con marcadores de activacion : CD30 +, CD15 +, CD 25 +, HLA-DR +, si bien el antigeno CD45 (antigeno leucocitario comun) y el CD20 (pan marcador B), son negativos, lo que ayuda a diferenciarlos del LNH. En el 85% de los casos de esclerosis nodular y celularidad mixta (y en una pequeña proporción de los otros subtipos), la célula de RS expresa niveles variables de CD30, aunque carece generalmente de expresión de CD15. Los procedimientos diagnósticos: emisión de positrones (EP), TAC y RM tienen la misma sensibilidad y especificidad, para determinar ganglios crecidos..
-TRATAMIENTO.
-RADIOTERAPIA. Dosis habituales.

-Campo implicado: de (35 -44 Gy) a (150-200 Gy), por fracción diaria (5 dias x semana)
-Campos no implicados : profiláctica de (30-35 Gy).
Definición de los campos:
Manto : cervical, supraclavicular, infraclavicular, mediastino y ganglios hiliares
Paraaórtico: bazo y pediculo esplénico.
Manto + paraaórtico : irradiación linfoide subtotal
Paraaórtico + pélvico :Y, invertida.
Manto + Y, Invertida : irradiación linfoide total.
-QUIMIOTERAPIA
-MOOP (mecloretamina, vincristina, procarbazina, prednisona)
-44% de los pacientes con enfermedad avanzada, libres de enfermedad, 20 años después
-AVBD (adriamicina, bleomicina, vinblastina y DTIC)
Eficáz en fallos de MOPP. Alternada con MOOP= regimenes alternantes o, hibridos
-REGIMENES ALTERNATIVOS, Empleados en enfermedad avanzada, incorporando G-CSF, para mitigar mielotoxicidad, por altas dosis.
-BEACOPP (bleomicina, etoposido, doxorubicina, ciclofosfamida, vincristina, procarbazina, prednisona
-Stanford V.
-ENFERMEDAD FAVORABLE, ESTADIO LIMITADO
-Estadio :IA/IIA, supradiafragmatica sin masas tumorales: radioterapia (manto o invertida) : 95-100 %, de supervivencia a los 5 años. Otra opción es administrar 3 ciclos de AVBD, continuada con radioterapia de campos afectados.
-ESTADIO LIMITADO, LOCALMENTE AVANZADA
Estadio : IA/IIA, con masa mediastinica voluminosa, (mas de 1/3, del diametro toracico abarcado por el tumor), caracterizada por presentar altas tasas de recaida con radioterapia, necesita de una modalidad de tratamiento combinada (80% de supervivencia libre de enfermedad, en comparación con el 50 %, con radioterapia sola). Para esta afección es mejor emplear combinaciones de radioterapia y quimioterapia. Afectación de ninguno o, solo un sitio extranodal : Irradiación en campo extendido (linfoide subtotal), como tratamiento de elección (80 %, libre de enfermedad a los 15 años). La quimioterapia combinada es curativa en 75-80%, de casos. Aunque el tratamiento del estadio IB/ IIB, sigue siendo controvertido, suele implicar el uso de quimioterapia o una modalidad combinada, dado que se le considera enfermedad diseminada.
-ENFERMEDAD AVANZADA
-La terapia en estadio IIIA, esta en debate. El problema es ¿la radioterapia implica irradiacion ganglionar total o es preferible la quimioterapia sistemica?. -La quimioterapia sistémica tambien esta en debate. ¿MOOP frente a AVBD, MOPP/AVBD o, nuevos regimenes : :BEACOPP, Stanford V. Parece que AVBD es preferible a hibridos, debido a su menor toxicidad. No obstante, el pronóstico mejora empleando: BEACOPP. En la actualidad se trata con radioterapia total a los enfermos con poca masa tumoral y con qumioterapia al resto. Para los estadios : IIIB y IV, se prefiere multiquimioterapia.
-ENFERMEDAD RECURRENTE
Las recidivas locales, se tratan con radioterapia. Para las recaidas sistemicas, si se empleo MOPP y la remision duro 1 año, emplear AVBD (40% de respuestas).-Recaida tras radioterapia: la qumioterapia logra una excelente tasa de curación, empleando 7 farmacos o mas. Si se emplearon terapias hibridas o alternantes -y hay fracaso- lo mejor es recurrir a trasplantes autologos de medula osea (40%, de sobrevida a los 5 años), o de celulas precursoras perifericas.
-COMPLICACIONES
-Neoplasias secundarias: LMA, mielodisplasia (1-10%, en 10 años), dependiente de dosis acumuladas de alquilantes. Riesgo de Linfoma agresivo difuso de células B, cánceres sólidos: pulmón, estómago, hueso, tejidos blandos (18% a los 15 años), dependiendo del grado de exposición a la quimioterapia. Puede desarrollarse una enfermedad cardiaca en receptores de radiación mediastinica. Es frecuente la infertilidad, tras tratamiento con MOPP. Puede aparecer una neumonitis por radiación, dependiendo de la dosis recibida por por el pulmon. Se producen anomalias de funcion tiroidea en cerca del 30% de los pacientes, tras irradiación tipo manto.
Labels: Linfoma de Hodgkin's
posted by Victor Mechán Mendez @ 4:17 PM
0 comments
![]()










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}